Mechanism of action
Odiparcil’s (IVA336) unique mechanism helps to reduce the GAG load in cells by presenting itself as an alternative substrate to galactosyl transferase, the enzyme initiating GAG synthesis. It is hypothesized that by shifting the synthesis of endogenous dermatan sulfate (DS) and chondroitin sulfate (CS) toward the synthesis of soluble (odiparcil-bound) DS and CS which can be eliminated via urine, odiparcil could reduce the cellular accumulation of non-metabolized endogenous substrates, the underlying cause of mucopolysaccharidoses.

Preclinical in vitro and in vivo efficacy data
In arylsulfatase B mutant mice, a relevant model for MPS VI, odiparcil showed to increase urinary GAG levels in a dose-dependent manner, in line with the mechanism described above. In addition there were consistent reductions in GAG levels in tissues and organs relevant to the human disease. Odiparcil decreased GAG accumulation in cornea and restored corneal structure and decreased GAG accumulation in cartilage which led to increased mobility. This is particularly important as these organs are affected in MPS VI but where current treatment is less effective. Distribution and exposure data generated in mice and rats indicate that the concentration levels reached in the heart, bones, cartilage and eyes are in the range or above the IC50 value of odiparcil for the reduction of intracellular GAG content.
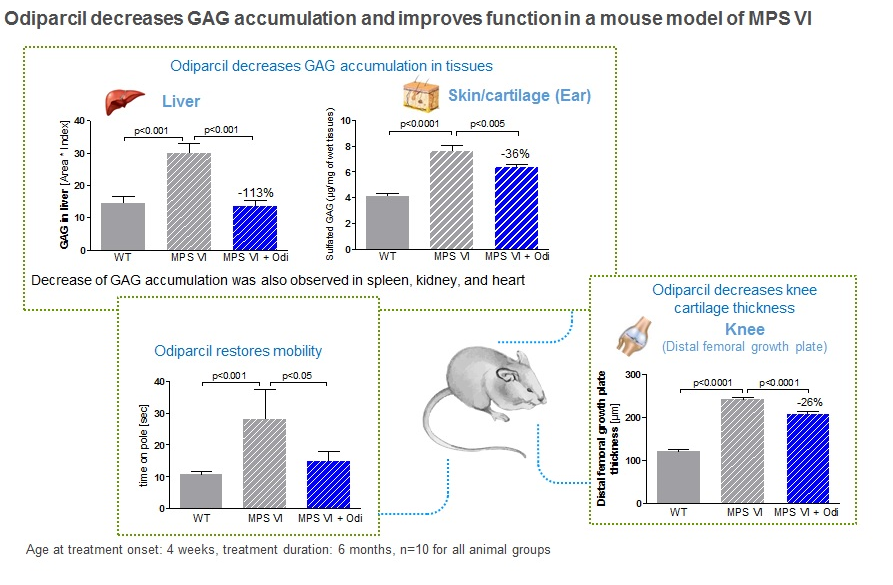
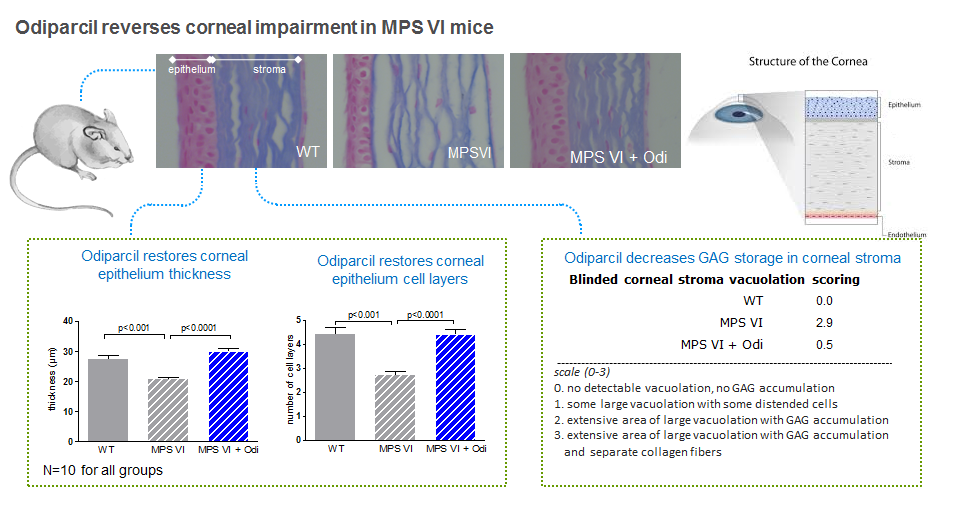
Odiparcil also decreased the intracellular levels of GAG in leukocytes in vivo in the MPSVI mouse model as well as in fibroblasts in vitro in MPS VI patient and healthy donor cells.
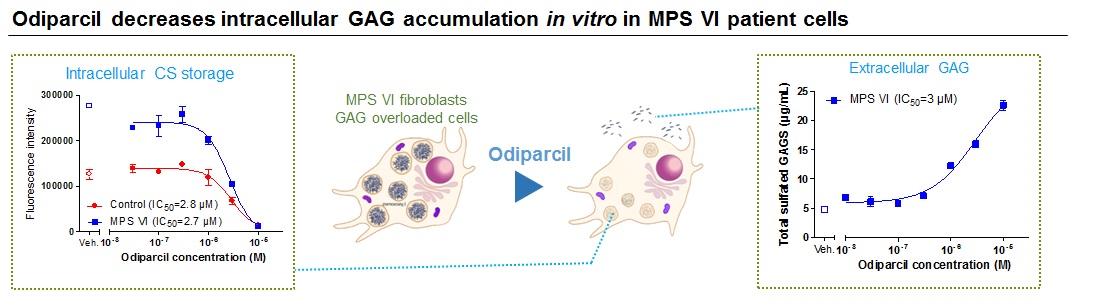
Source: Inventiva S.A. proprietary data
Preclinical in vivo safety data
The observed pharmacological active dose of odiparcil in the animal model above corresponds to a concentration in vitro which leads to a reduction of intracellular GAG levels but not to their complete depletion. Accordingly, the observed pharmacological active dose results in an exposure that was shown to be safe in preclinical in vivo toxicology studies in mice, rats and monkeys (up to 52 weeks) and was also shown to be safe in adults during clinical studies (approx. 1900 healthy volunteers and patients exposed for up to 16 weeks).